Effects on HUVECs to the natural-source compound 1. Endothelial cells from different tissues have different gene expression patterns suggesting different physiological effects. HUVECs are macrovascular endothelial cells which do not have specific relevance to the microvascular endothelial cells that are present in retinal capillaries of the eye. Therefore, we tested SH11052 in HRECs, where it proved similarly potent as an antiproliferative molecule, albeit at higher GI50, consistent with the hypothesis that microvascular endothelial cells differ from macrovascular endothelial cells. SH-11052 blocks proliferative progression through DNA synthesis in both HUVECs and HRECs. This is consistent with the documented G2/M phase cell cycle arrest induced by 1 in HUVECs. We have also demonstrated the in vitro anti-angiogenic activity of 2 in a Matrigel HREC tube formation assay, similar to the effects of 1 in HUVECs. The novel anti-angiogenic mechanism of homoisoflavanones remains largely unexplored. In HUVECs, compound 1 induced expression of p21WAF1, an inhibitor of the cyclindependent kinase Cdc2, which in turn is BIBW2992 downregulated by compound 1. Homoisoflavanone 1 also blocked prostaglandin synthesis from arachidonic acid in a microsome assay, without marked effects on WZ4002 function of cyclooxygenases 1 and 2 as purified enzymes. In keratinocytes, compound 1 inhibited the nuclear translocation of NF-kB under ultraviolet light-induced inflammatory conditions, suggesting a role of the compound in modulating inflammatory signals in these cells. In this context, compound 1 also decreased phosphorylation of the MAPKs Jun N-terminal kinase, p38 MAPK, and ERK. We examined if the activity of SH-11052 in HRECs may likewise be mediated through modulation of inflammatory signals. As NF-kB is the principal mediator of inflammation induced signals, we monitored NF-kB activation upon TNF-a stimulation in the absence or presence of compound 2 in HRECs. NF-kB is a transcription factor sequestered in the cytoplasm by association with IkB-a protein. Upon TNF-a stimulation, IkB-a is phosphorylated and degraded by the proteasome, releasing free NF-kB. The free NF-kB is then translocated into the nucleus and aids in the transcription of its target genes. Hence monitoring the protein levels of IkB-a and nuclear translocation of NF-kB upon TNF-a treatment are measures of the activation of the NF-kB pathway by inflammatory signals. Indeed, we show that IkBa degradation and nuclear translocation of NF-kB in HRECs are inhibited by compound 2. Furthermore, compound 2 also inhibited the expression of NF-kB inducible pro-angiogenic and pro-inflammatory genes, suggesting a role for compound 2 in the inhibition of inflammation-induced pro-angiogenic signaling in HRECs. SH-11052’s suppressive effects on expression of IL8 and PTGS2 in HRECs are consistent with the observed effects of the natural product 1 in keratinocytes. To our knowledge, we show for the first time an effect of a homoisoflavanone on the endothelial activation marker and NF-kB target, VCAM-1, and 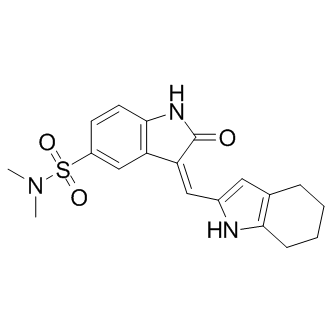 on the inflammatory marker CCL2. Thus, the data presented here are consistent with a function for compound 2 as an inhibitor of NFkB signaling in HRECs. NF-kB has previously been implicated in pathological ocular angiogenesis and we further confirmed that two known NF-kB pathway inhibitors, BAY 11-7082 and CAPE, had antiproliferative effects on HRECs. This assertion of an NF-kB-dependent role for compound 2 can integrate others’ findings regarding the activities of the related compound 1 in other cell types as well. bFGF can act by signaling through phospholipase Cc1, which activates protein kinase C a via diacylglycerol. In turn, PKCa binds and activates IKKa, which phosphorylates and inactivates I-kB.
on the inflammatory marker CCL2. Thus, the data presented here are consistent with a function for compound 2 as an inhibitor of NFkB signaling in HRECs. NF-kB has previously been implicated in pathological ocular angiogenesis and we further confirmed that two known NF-kB pathway inhibitors, BAY 11-7082 and CAPE, had antiproliferative effects on HRECs. This assertion of an NF-kB-dependent role for compound 2 can integrate others’ findings regarding the activities of the related compound 1 in other cell types as well. bFGF can act by signaling through phospholipase Cc1, which activates protein kinase C a via diacylglycerol. In turn, PKCa binds and activates IKKa, which phosphorylates and inactivates I-kB.
Blockade of this pathway would inhibit cellular responses to bFGF
Leave a reply
 associated online supplementary although the exact mechanism is still in investigation. Ubiquitinated VDAC1 and Drp1 will cause their associated mitochondria to be brought into autophagosomes and autolysosomes for degradation. Since a significant portion of Bcl-2 is associated with mitochondria and Parkin-mono-ubiquitinated Bcl2 is more stable, suppression of LRPPRC leads to decreases in levels of Parkin and Bcl-2 and activation of basal autophagy as we previous reported. Interestingly, Parkin itself is the substrate of its ligase activity. After auto-ubiquitination, Parkin gradually becomes depleted along Bcl-2 and ATG5-ATG12 conjugate in cells under long-term mitophagy stress. The drug-induced mitophagy stress is an artificially introduced pathological condition. Under normal physiological condition, it is unlikely that all of mitochondria in cells are simultaneously damaged. The drug-induced mitochondrial damages are so massive that autophagy/mitophagy machinery is incapable of handing so many damaged mitochondria immediately. This is possibly the reason that we observed a large
associated online supplementary although the exact mechanism is still in investigation. Ubiquitinated VDAC1 and Drp1 will cause their associated mitochondria to be brought into autophagosomes and autolysosomes for degradation. Since a significant portion of Bcl-2 is associated with mitochondria and Parkin-mono-ubiquitinated Bcl2 is more stable, suppression of LRPPRC leads to decreases in levels of Parkin and Bcl-2 and activation of basal autophagy as we previous reported. Interestingly, Parkin itself is the substrate of its ligase activity. After auto-ubiquitination, Parkin gradually becomes depleted along Bcl-2 and ATG5-ATG12 conjugate in cells under long-term mitophagy stress. The drug-induced mitophagy stress is an artificially introduced pathological condition. Under normal physiological condition, it is unlikely that all of mitochondria in cells are simultaneously damaged. The drug-induced mitochondrial damages are so massive that autophagy/mitophagy machinery is incapable of handing so many damaged mitochondria immediately. This is possibly the reason that we observed a large