In FA transport and the response to a fat diet and specific defect in FA uptake in the proximal intestine of CD362/2 mice is associated with reduced incorporation of FA in TG and a diminished TG secretion. This concept was however AP24534 challenged. Published observations have shown that CD36 genetic deletion does not affect intestinal lipid uptake and the efficient participation of CD36 in LCFA intestinal uptake was questioned. It was suggested that CD36 functions as a FA sensor and stimulates events that control FA metabolism rather than being directly involved in the lipid transit. In any case, our findings show that small inhibitors of the CD36 binding functions can significantly reduce the postprandial BAY 73-4506 hypertriglyceridemia which follows a gastric olive challenge. Again, when compared to a complete deletion of the gene, which favor redundant mechanisms, a partial inhibition of CD36 functions may have different consequences. Our findings demonstrate that a selective down regulation of CD36 in the intestine reduces lipid intake and is beneficial to postprandial hypertriglyceridemia. In conclusion, CD36 is generally recognized as an important lipid and FA receptor which plays a role in the metabolic syndrome and its associated cardiac events. The pleiotropic activity and the various molecular associations of this scavenger in different cells and tissues have however questioned its potential as a safe therapeutic target. Different published observations have indeed suggested that CD36 down regulation might not been beneficial due to redundant mechanisms or potential toxicity. The present study shows that it is possible to identify small molecules that can block the CD36 binding and uptake functions and that such antagonism can reduce atherosclerosis, postprandial hypertriglyceridemia and be beneficial for type II diabetes. Particularly, elevated postprandial hypertriglyceridemia is a metabolic parameter which is now recognized to be strongly associated with cardiovascular events and is independent of traditional cardiovascular risk factors. Thus, CD36 might represent an attractive therapeutic target. Its expression can be induced by a broad spectrum of growth factors, hormones, or stress stimuli, and it is associated with various chronic conditions. Studies in mice have revealed that Mig-6 is required for skin morphogenesis and lung development and that it plays an important role in maintaining joint homeostasis. As a cytoplasmic scaffolding adaptor, MIG-6 has several important protein-protein interaction motifs that may mediate interaction with signaling molecules downstream of receptor tyrosine kinases. One of the most prominent roles of MIG-6 in regulating signal transduction comes from its ability to directly interact with epidermal growth factor receptor 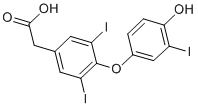 and other ErbB family members, inhibiting their phosphorylation and downstream signaling in a negative feedback fashion. MIG-6 can be induced by hepatocyte growth factor and functions as a negative feedback regulator of HGF-MET signaling, indicating that it has broad role as a signal checkpoint for modulating activated RTK pathways in a timely manner. The evidence that MIG-6 is a tumor suppressor gene is compelling. It is located in chromosome 1p36, a locus that frequently has loss of heterozygosity in several human cancers including lung cancer, melanoma, and breast cancer. Indeed, down-regulation or loss of MIG-6 expression has been reported in cancers and is often associated with poor prognosis. MIG-6 down-regulation in non-small cell lung cancer is associated with increased EGFR signaling and poorly differentiated cancer, while loss of its expression in ErbB2-amplified breast carcinoma renders the cancer cells more resistant to Herceptin, the neutralizing antibody against ErbB2.
and other ErbB family members, inhibiting their phosphorylation and downstream signaling in a negative feedback fashion. MIG-6 can be induced by hepatocyte growth factor and functions as a negative feedback regulator of HGF-MET signaling, indicating that it has broad role as a signal checkpoint for modulating activated RTK pathways in a timely manner. The evidence that MIG-6 is a tumor suppressor gene is compelling. It is located in chromosome 1p36, a locus that frequently has loss of heterozygosity in several human cancers including lung cancer, melanoma, and breast cancer. Indeed, down-regulation or loss of MIG-6 expression has been reported in cancers and is often associated with poor prognosis. MIG-6 down-regulation in non-small cell lung cancer is associated with increased EGFR signaling and poorly differentiated cancer, while loss of its expression in ErbB2-amplified breast carcinoma renders the cancer cells more resistant to Herceptin, the neutralizing antibody against ErbB2.
In glioblastoma MIG-6 is identified as a single gene within the most commonly deleted
Leave a reply
 rare, three mutations in the MIG-6 gene have been identified in human lung cancer and one in neuroblastoma. Further evidence supporting MIG-6 as a tumor suppressor gene arose from mouse studies; Mig-6deficient mice are prone to develop epithelial hyperplasia or tumors in organs including the lung, skin, uterus, gallbladder, and bile duct. Epigenetic alteration, one of the most well-known mechanisms leading to inactivation of a tumor suppressor gene, can result from DNA methylation or histone deacetylation in the gene’s promoter. Given that down-regulation of MIG-6 is frequently observed in many human cancers, we asked whether MIG-6 expression was affected by DNA methylation and histone deacetylation. Here, we show that the MIG-6
rare, three mutations in the MIG-6 gene have been identified in human lung cancer and one in neuroblastoma. Further evidence supporting MIG-6 as a tumor suppressor gene arose from mouse studies; Mig-6deficient mice are prone to develop epithelial hyperplasia or tumors in organs including the lung, skin, uterus, gallbladder, and bile duct. Epigenetic alteration, one of the most well-known mechanisms leading to inactivation of a tumor suppressor gene, can result from DNA methylation or histone deacetylation in the gene’s promoter. Given that down-regulation of MIG-6 is frequently observed in many human cancers, we asked whether MIG-6 expression was affected by DNA methylation and histone deacetylation. Here, we show that the MIG-6 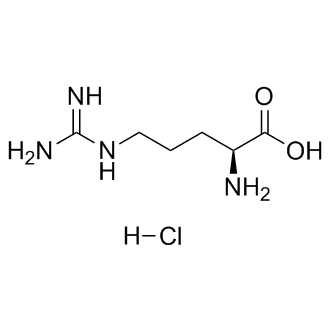 for response to chemotherapy, especially gemcitabine therapy, a first line treatment for pancreatic cancer. Our findings that a specific Akt inhibitor can reverse resistance to gemcitabine in FKBP5 knockdown cells and xenografts indicate that FKBP5 levels might be used to stratify patients into different treatment arms, such as gemcitabine or gemcitabine plus an Akt inhibitor. Future clinical studies will be needed to test this hypothesis. In addition, the mechanisms underlying differences between the effects of PI3K inhibition, mTOR inhibition and Akt inhibition in combination with gemcitabine need to be explored further. PI3K activation causes phosphatidylinositol-3,4,5-triphosphate dependent membrane localization of Akt and PDK1, in which the latter can phosphorylate Akt 308. Therefore, the inhibition of PI3K might have less effect on 473 phosphorylation. Rapamycin can potentially
for response to chemotherapy, especially gemcitabine therapy, a first line treatment for pancreatic cancer. Our findings that a specific Akt inhibitor can reverse resistance to gemcitabine in FKBP5 knockdown cells and xenografts indicate that FKBP5 levels might be used to stratify patients into different treatment arms, such as gemcitabine or gemcitabine plus an Akt inhibitor. Future clinical studies will be needed to test this hypothesis. In addition, the mechanisms underlying differences between the effects of PI3K inhibition, mTOR inhibition and Akt inhibition in combination with gemcitabine need to be explored further. PI3K activation causes phosphatidylinositol-3,4,5-triphosphate dependent membrane localization of Akt and PDK1, in which the latter can phosphorylate Akt 308. Therefore, the inhibition of PI3K might have less effect on 473 phosphorylation. Rapamycin can potentially 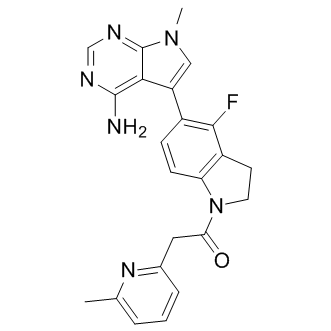 in MOLOC. Partial charges for the co-factor were calculated using AMSOL. Spheres as matching points for docking were placed around the cytidine heterocycle of the bound substrate. The sphere set defining the buried region of the binding site was generated around the whole substrate and cofactor. Grids to store information about excluded volumes, electrostatic and van der Waals potential, and solvent occlusion were calculated as described earlier. Each docking pose which did not place any atoms in areas occupied by the receptor was scored for electrostatic and van der Waals complementarity and
in MOLOC. Partial charges for the co-factor were calculated using AMSOL. Spheres as matching points for docking were placed around the cytidine heterocycle of the bound substrate. The sphere set defining the buried region of the binding site was generated around the whole substrate and cofactor. Grids to store information about excluded volumes, electrostatic and van der Waals potential, and solvent occlusion were calculated as described earlier. Each docking pose which did not place any atoms in areas occupied by the receptor was scored for electrostatic and van der Waals complementarity and 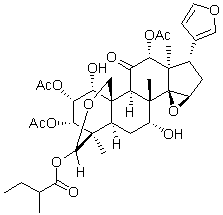 This is not surprising since Golgi and ER, which get disrupted during cell lysis, contain high amounts of both complete and partly synthesized glycans and these can alter the proportion of a particular glycan group. Each of the two glycan release methods was repeated six times to estimate their reproducibility. For nearly all glycan peaks, we observed higher coefficients of variation when glycome was analyzed from cell lysates. Especially high experimental variation was observed in highly branched glycan structures, which are important for the regulation of membrane half-life of many receptors. This increased variation of complex structures from experiment to experiment probably reflects their small contribution to the total glycome. Since glycans function as regulators of the activity of membrane proteins, many of them are also attached to proteins on the luminal side of various cytoplasmic vesicles, including the Golgi apparatus where posttranslational glycan processing occurs. Homogenization of cells results in mixing of these two compartments, containing physiologically separate fractions of glycans, and consequently leads to masking of relatively subtle, but functionally important, differences in the cell membrane Nglycome. Therefore, we believe that the analysis of Nglycome from embedded cells results in improved analytical precision due to elimination of the homogenization step from the procedure. Based on these observations we decided to further exploit the method of glycan analysis from embedded cells. We validated this hypothesis by mannosidase treatment, which resulted in
This is not surprising since Golgi and ER, which get disrupted during cell lysis, contain high amounts of both complete and partly synthesized glycans and these can alter the proportion of a particular glycan group. Each of the two glycan release methods was repeated six times to estimate their reproducibility. For nearly all glycan peaks, we observed higher coefficients of variation when glycome was analyzed from cell lysates. Especially high experimental variation was observed in highly branched glycan structures, which are important for the regulation of membrane half-life of many receptors. This increased variation of complex structures from experiment to experiment probably reflects their small contribution to the total glycome. Since glycans function as regulators of the activity of membrane proteins, many of them are also attached to proteins on the luminal side of various cytoplasmic vesicles, including the Golgi apparatus where posttranslational glycan processing occurs. Homogenization of cells results in mixing of these two compartments, containing physiologically separate fractions of glycans, and consequently leads to masking of relatively subtle, but functionally important, differences in the cell membrane Nglycome. Therefore, we believe that the analysis of Nglycome from embedded cells results in improved analytical precision due to elimination of the homogenization step from the procedure. Based on these observations we decided to further exploit the method of glycan analysis from embedded cells. We validated this hypothesis by mannosidase treatment, which resulted in