The plasma glycome which revealed very high variability in the composition of plasma glycome in the population and identified individuals having significantly aberrant glyco-phenotypes, some of which could be BAY-60-7550 associated with specific diseases. To test potential therapeutic usefulness of epigenetic inhibitors TSA, sodium butyrate and zebularine in inducing a reversal of undesired glyco-phenotypes, we developed an HPLCbased method for the determination of glycan structures from cells embedded in polyacrylamide gels. In addition, we specifically investigated the preservation of altered glycan profiles over a prolonged period of time in a drug-free environment. Our results emphasize the importance of epigenetic control in the regulation of N-glycosylation, but also suggest the stability of complex biosynthetic pathways responsible for the establishment of glycan profiles in human cells in culture. Indeed, it has been shown that the overexpression of glycoproteins with oligomannose N-glycans at the cell surface induces proliferative and adhesive properties of cancer cells and potentially facilitates loss of contact inhibition, a hallmark feature of majority of cancer cells. Since glycans can also be obtained following disruption of cellular integrity, we decided to compare the methodological precision of N-glycan analysis from embedded cells to the total Nglycome obtained by the analysis of HeLa cell lysates. The observed glycan profiles were somewhat different, with particular glycan groups being selectively more abundant in the lysates or the embedded cell fraction. 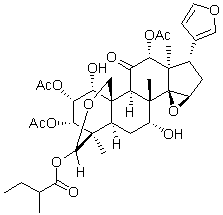 This is not surprising since Golgi and ER, which get disrupted during cell lysis, contain high amounts of both complete and partly synthesized glycans and these can alter the proportion of a particular glycan group. Each of the two glycan release methods was repeated six times to estimate their reproducibility. For nearly all glycan peaks, we observed higher coefficients of variation when glycome was analyzed from cell lysates. Especially high experimental variation was observed in highly branched glycan structures, which are important for the regulation of membrane half-life of many receptors. This increased variation of complex structures from experiment to experiment probably reflects their small contribution to the total glycome. Since glycans function as regulators of the activity of membrane proteins, many of them are also attached to proteins on the luminal side of various cytoplasmic vesicles, including the Golgi apparatus where posttranslational glycan processing occurs. Homogenization of cells results in mixing of these two compartments, containing physiologically separate fractions of glycans, and consequently leads to masking of relatively subtle, but functionally important, differences in the cell membrane Nglycome. Therefore, we believe that the analysis of Nglycome from embedded cells results in improved analytical precision due to elimination of the homogenization step from the procedure. Based on these observations we decided to further exploit the method of glycan analysis from embedded cells. We validated this hypothesis by mannosidase treatment, which resulted in Tubulin Acetylation Inducer almost complete disappearance of the corresponding chromatographic peaks, thus confirming the contribution of free oligomannose glycans to the pool of glycans released by PNGase F from glycoproteins associated with the cell membrane.
This is not surprising since Golgi and ER, which get disrupted during cell lysis, contain high amounts of both complete and partly synthesized glycans and these can alter the proportion of a particular glycan group. Each of the two glycan release methods was repeated six times to estimate their reproducibility. For nearly all glycan peaks, we observed higher coefficients of variation when glycome was analyzed from cell lysates. Especially high experimental variation was observed in highly branched glycan structures, which are important for the regulation of membrane half-life of many receptors. This increased variation of complex structures from experiment to experiment probably reflects their small contribution to the total glycome. Since glycans function as regulators of the activity of membrane proteins, many of them are also attached to proteins on the luminal side of various cytoplasmic vesicles, including the Golgi apparatus where posttranslational glycan processing occurs. Homogenization of cells results in mixing of these two compartments, containing physiologically separate fractions of glycans, and consequently leads to masking of relatively subtle, but functionally important, differences in the cell membrane Nglycome. Therefore, we believe that the analysis of Nglycome from embedded cells results in improved analytical precision due to elimination of the homogenization step from the procedure. Based on these observations we decided to further exploit the method of glycan analysis from embedded cells. We validated this hypothesis by mannosidase treatment, which resulted in Tubulin Acetylation Inducer almost complete disappearance of the corresponding chromatographic peaks, thus confirming the contribution of free oligomannose glycans to the pool of glycans released by PNGase F from glycoproteins associated with the cell membrane.
A tiny fraction of remaining peaks could as well predominantly represent glycan structures transported to the cell surface
Leave a reply
Ginsenoside-Rh2.png) that the therapeutic agent mizoribine monophosphate
that the therapeutic agent mizoribine monophosphate  BEZ235-induced accumulation of cells at G0/G1 phase may therefore reduce the therapeutic advantage of concurrent therapy with topoisomerase inhibitors, explaining the unfavorable combination effects of BEZ235 with irinotecan and etoposide. Daily treatment of BEZ235 significant retarded 8505C xenograft tumor growth during the therapeutic period. The inhibitory effect was less prominent after the discontinuation of therapy, suggesting that prolonged treatment may be necessary to maintain therapeutic efficacy. BEZ235 significantly degraded caspase-3 in 8505C xenograft tumors, indicating this compound may induce apoptosis in vivo. After discontinuation of BEZ235, the volume of 8505C xenograft tumors subsequently increased. This enlargement of tumor following cessation of therapy might be due to normalization of cell size and resumption of cell proliferation, as mTOR and its downstream proteins S6 Kinase 1 and 4E-BP1 play pivotal roles in this respect. No significant weight loss or illness was observed during study period, suggesting that this therapy may have a promising safety profile. A prior report demonstrated that the presence of genetic alterations of PTEN, PIK3CA and AKT1 correlated well with sensitivity to an AKT inhibitor, but had weaker correlations with an mTOR inhibitor. This discrepency suggests that mTOR activity does not depend solely on PI3K/AKT activity. The reported data of genetic alterations in 8 thyroid cancer cell lines was summarized. Limited genetic changes on RAS/RAF/ERK and PI3K/mTOR pathways were identified.
BEZ235-induced accumulation of cells at G0/G1 phase may therefore reduce the therapeutic advantage of concurrent therapy with topoisomerase inhibitors, explaining the unfavorable combination effects of BEZ235 with irinotecan and etoposide. Daily treatment of BEZ235 significant retarded 8505C xenograft tumor growth during the therapeutic period. The inhibitory effect was less prominent after the discontinuation of therapy, suggesting that prolonged treatment may be necessary to maintain therapeutic efficacy. BEZ235 significantly degraded caspase-3 in 8505C xenograft tumors, indicating this compound may induce apoptosis in vivo. After discontinuation of BEZ235, the volume of 8505C xenograft tumors subsequently increased. This enlargement of tumor following cessation of therapy might be due to normalization of cell size and resumption of cell proliferation, as mTOR and its downstream proteins S6 Kinase 1 and 4E-BP1 play pivotal roles in this respect. No significant weight loss or illness was observed during study period, suggesting that this therapy may have a promising safety profile. A prior report demonstrated that the presence of genetic alterations of PTEN, PIK3CA and AKT1 correlated well with sensitivity to an AKT inhibitor, but had weaker correlations with an mTOR inhibitor. This discrepency suggests that mTOR activity does not depend solely on PI3K/AKT activity. The reported data of genetic alterations in 8 thyroid cancer cell lines was summarized. Limited genetic changes on RAS/RAF/ERK and PI3K/mTOR pathways were identified. as stroke, myocardial
as stroke, myocardial