HhAntag691 moa Furthermore, PAbN has been reported to have no inhibitory effect on efflux of ethidium bromide, which is also known as a substrate of ABC-type transporter. What is more, fluorescence of these compounds was not as strong as that of fluorescein, and thus they are not considered to be suitable for the assay in the microfluidic channel. The role of efflux 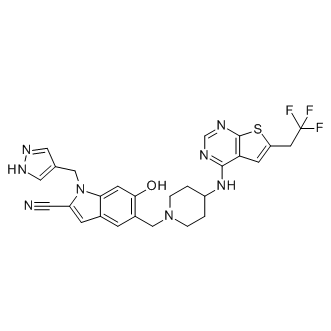 pumps on FDG hydrolysis in E. coli was not fully understood when we chose FDG as a substrate for the assay at the beginning of this study. FDG was defined as a substrate of RND pumps because it was easily hydrolyzed in pump deletion mutants compared with wild-type cells. If FDG was not a substrate of pumps, it will be difficult to discriminate DB from wild-type. Fluorescein was also defined as a substrate of pumps from its accumulation in DC cells. Furthermore, from the results of complete blockage of FDG hydrolysis by protonophore CCCP addition in all of the strains including DC, FDG was defined to be actively imported into the cytoplasm. The lactose permease LacY is not a FDG permease because lacY deletion from DlacI mutants had no effect on FDG hydrolysis in the mutants, and we have yet to identify a FDG permease. Figure 12 shows the proposed AZ 960 mechanism of FDG hydrolysis in E. coli. In wild-type cells, FDG is hardly imported into cytoplasm because FDG is exported by AcrB from periplasm before it is trapped by permease. Rate of FDG influx will increase depending on a concentration of FDG in periplasm until it reach maximum rate. Moderate inhibition of the pumps causes FDG influx and an efflux of fluorescein from the cells by the remaining activity of the pumps, and full inhibition of the pumps results in fluorescein accumulation in cells, similar to what is observed in DC cells. A real pump inhibitor without any effect on bacterial membrane will increase fluorescence in wild-type cell by concentration dependent manner and will cause increased accumulation of fluorescence in the cells. In fact, we detected moderately increasing fluorescence and increased accumulation of fluorescence in DBC/pABM according to increasing concentration of D13-9001. In contrast, peameabilization of outer membrane will cause leakage of fluorescein from DC cells, and peameabilization of inner membrane will efficiently increase FDG influx and fluorescein production to release it from cells with or without pumps. By the FDG assay, it is easy to detect outer membrane peameabilization by disappearance of fluorescein accumulation in DC cells, and inner membrane peameabilization by increased fluorescence especially in the medium of pump deletion mutants. Determination of efflux pump inhibition activity via the typical methods of measuring the influx or efflux of some substrates by their fluorescence with a plate reader makes it difficult to exclude the effect of outer membrane peameabilizing activity. In addition, accurate estimation of FDG hydrolysis through monitoring fluorescence with a plate-reader is impractical because the total fluorescence of fluorescein is higher when diffused in the medium than when accumulated in the cells, and fluorescence determined by a plate reader was usually higher in DB than in DC. Therefore, fluorescence determined by a plate reader does not accurately correlate with the amount of fluorescein produced, and it is difficult to estimate the inhibitory effect on pumps with FDG by a plate reader. The microfluidic channel method enables discrimination of pure efflux pump inhibition from membrane permeabilization.
pumps on FDG hydrolysis in E. coli was not fully understood when we chose FDG as a substrate for the assay at the beginning of this study. FDG was defined as a substrate of RND pumps because it was easily hydrolyzed in pump deletion mutants compared with wild-type cells. If FDG was not a substrate of pumps, it will be difficult to discriminate DB from wild-type. Fluorescein was also defined as a substrate of pumps from its accumulation in DC cells. Furthermore, from the results of complete blockage of FDG hydrolysis by protonophore CCCP addition in all of the strains including DC, FDG was defined to be actively imported into the cytoplasm. The lactose permease LacY is not a FDG permease because lacY deletion from DlacI mutants had no effect on FDG hydrolysis in the mutants, and we have yet to identify a FDG permease. Figure 12 shows the proposed AZ 960 mechanism of FDG hydrolysis in E. coli. In wild-type cells, FDG is hardly imported into cytoplasm because FDG is exported by AcrB from periplasm before it is trapped by permease. Rate of FDG influx will increase depending on a concentration of FDG in periplasm until it reach maximum rate. Moderate inhibition of the pumps causes FDG influx and an efflux of fluorescein from the cells by the remaining activity of the pumps, and full inhibition of the pumps results in fluorescein accumulation in cells, similar to what is observed in DC cells. A real pump inhibitor without any effect on bacterial membrane will increase fluorescence in wild-type cell by concentration dependent manner and will cause increased accumulation of fluorescence in the cells. In fact, we detected moderately increasing fluorescence and increased accumulation of fluorescence in DBC/pABM according to increasing concentration of D13-9001. In contrast, peameabilization of outer membrane will cause leakage of fluorescein from DC cells, and peameabilization of inner membrane will efficiently increase FDG influx and fluorescein production to release it from cells with or without pumps. By the FDG assay, it is easy to detect outer membrane peameabilization by disappearance of fluorescein accumulation in DC cells, and inner membrane peameabilization by increased fluorescence especially in the medium of pump deletion mutants. Determination of efflux pump inhibition activity via the typical methods of measuring the influx or efflux of some substrates by their fluorescence with a plate reader makes it difficult to exclude the effect of outer membrane peameabilizing activity. In addition, accurate estimation of FDG hydrolysis through monitoring fluorescence with a plate-reader is impractical because the total fluorescence of fluorescein is higher when diffused in the medium than when accumulated in the cells, and fluorescence determined by a plate reader was usually higher in DB than in DC. Therefore, fluorescence determined by a plate reader does not accurately correlate with the amount of fluorescein produced, and it is difficult to estimate the inhibitory effect on pumps with FDG by a plate reader. The microfluidic channel method enables discrimination of pure efflux pump inhibition from membrane permeabilization.
It may be difficult to detect the effect of a specific inhibitor on either of them
Leave a reply
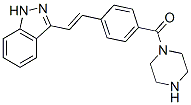 of OVs in vivo and that further clinical evaluation of this possibility is warranted. We therefore reasoned that if the AA target was the cytosolic CyP-A, the drug could also act on other members of this protein family. Indeed, such a pleiotropic effect is well-characterized for CsA, as CsA also targets the mitochondria-restricted CyP-D. CyP-D displays an important role in the cell response to a variety of noxious stimuli, as it modulates a channel located in the inner mitochondrial membrane, the permeability transition pore, whose prolonged opening irreversibly commits cells to death. PTP dysregulation is emerging as a common feature in a variety of pathologies endowed with either an excess of cell death, such as neurodegenerative disease or muscular dystrophies, or with an aberrant hyperactivation of survival pathways, as in cancer. CsA inhibits PTP opening through binding to CyP-D. Therefore, it constitutes an interesting molecule for the treatment of degenerative diseases. Nonetheless, due to its immunosuppressant activity, to its side effects and to its inability to pass the blood-brain barrier, CsA analogues with a higher selectivity for CyP-D are under intense scrutiny. Here we demonstrate that, similar to CsA, AA targets CyP-D leading to PTP inhibition and to cell protection from insults that cause pore opening. AA could be exploited as a lead compound for a new class of PTP-inhibiting drugs. The identification of PTP targeting drugs is a highly desirable result, as the PTP is involved in a wide range of diseases. By the use of CsA or of CyP-D knock-out animals it was established that dysregulated.
of OVs in vivo and that further clinical evaluation of this possibility is warranted. We therefore reasoned that if the AA target was the cytosolic CyP-A, the drug could also act on other members of this protein family. Indeed, such a pleiotropic effect is well-characterized for CsA, as CsA also targets the mitochondria-restricted CyP-D. CyP-D displays an important role in the cell response to a variety of noxious stimuli, as it modulates a channel located in the inner mitochondrial membrane, the permeability transition pore, whose prolonged opening irreversibly commits cells to death. PTP dysregulation is emerging as a common feature in a variety of pathologies endowed with either an excess of cell death, such as neurodegenerative disease or muscular dystrophies, or with an aberrant hyperactivation of survival pathways, as in cancer. CsA inhibits PTP opening through binding to CyP-D. Therefore, it constitutes an interesting molecule for the treatment of degenerative diseases. Nonetheless, due to its immunosuppressant activity, to its side effects and to its inability to pass the blood-brain barrier, CsA analogues with a higher selectivity for CyP-D are under intense scrutiny. Here we demonstrate that, similar to CsA, AA targets CyP-D leading to PTP inhibition and to cell protection from insults that cause pore opening. AA could be exploited as a lead compound for a new class of PTP-inhibiting drugs. The identification of PTP targeting drugs is a highly desirable result, as the PTP is involved in a wide range of diseases. By the use of CsA or of CyP-D knock-out animals it was established that dysregulated.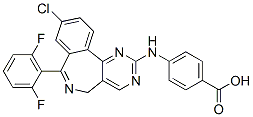 6, meaning that the spread of H1N1 influenza virus has become a serious global pandemic. It was anticipated that a stronger outbreak might occur in the coming winter. The even worse news is that cases were reported that several strains of H1N1 influenza A viruses were resistant to oseltamivir. Although an influenza virus only possesses eight genes, its simplicity has not stopped it from wreaking havoc on human beings for centuries. “The only thing predictable about influenza is its unpredictability”. Influenza A virus has the ability to undergo changes by the mechanisms of antigenic drift and shift, resulting in new evolving virus strains, which may be extremely toxic and drug-resistant. Given that influenza shifts may occur every 20�C30 years, the danger of future influenza A pandemics highlights the need to develop more effective drugs. The threat of an impending influenza pandemic, possibly through the mutations of the present avian strain H5N1 or swine strain H1N1, has triggered a global effort to develop more effective antivirus drugs.
6, meaning that the spread of H1N1 influenza virus has become a serious global pandemic. It was anticipated that a stronger outbreak might occur in the coming winter. The even worse news is that cases were reported that several strains of H1N1 influenza A viruses were resistant to oseltamivir. Although an influenza virus only possesses eight genes, its simplicity has not stopped it from wreaking havoc on human beings for centuries. “The only thing predictable about influenza is its unpredictability”. Influenza A virus has the ability to undergo changes by the mechanisms of antigenic drift and shift, resulting in new evolving virus strains, which may be extremely toxic and drug-resistant. Given that influenza shifts may occur every 20�C30 years, the danger of future influenza A pandemics highlights the need to develop more effective drugs. The threat of an impending influenza pandemic, possibly through the mutations of the present avian strain H5N1 or swine strain H1N1, has triggered a global effort to develop more effective antivirus drugs.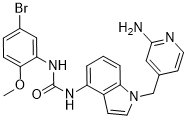 fact represent the cause rather than the consequence of the adaptive pressure. By considering that the glycolytic switch typical of cancer cells is acquired at the very onset of carcinogenesis, the idea arose that alterations in the glycolytic pathway may predispose cells to malignant transformation. Selective advantages for the transformed cells could result from various features. For instance, it is known that hypoxia-inducible factor-1 greatly stimulates the expression of glucose and monocarboxylate transporters, glycolytic enzymes and induces a down regulation in pyruvate dehydrogenase complex.
fact represent the cause rather than the consequence of the adaptive pressure. By considering that the glycolytic switch typical of cancer cells is acquired at the very onset of carcinogenesis, the idea arose that alterations in the glycolytic pathway may predispose cells to malignant transformation. Selective advantages for the transformed cells could result from various features. For instance, it is known that hypoxia-inducible factor-1 greatly stimulates the expression of glucose and monocarboxylate transporters, glycolytic enzymes and induces a down regulation in pyruvate dehydrogenase complex.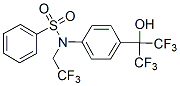 particularly sensitive when survival pathway inhibitors are combined with mitotic inhibitors. Moreover, combination of bortezomib with mitotic inhibitors are currently in clinical trials for the treatment of non-small-cell lung carcinoma and other solid tumors. Thus, we hypothesized that a strategy based on the combined treatment with bortezomib and mitotic inhibitors for the treatment of Bcr-Abl-positive leukemias may be promising. Especially important might be to determine the effectiveness of this strategy in TKIs-resistant Bcr-Abl-positive cases. Paclitaxel, a mitotic inhibitor drug acting by stabilization of microtubules, is FDA approved for the treatment of lung, ovarian, breast cancers and advanced forms of Kaposi’s sarcoma. Paclitaxel is now in clinical trials for the treatment of CML. However, to our knowledge, there are no clinical trials or published studies employing the combined bortezomib and paclitaxel regimen for the treatment of Bcr-Ablpositive CML. Such a combination, if synergistic in inducing apoptosis in Bcr-Abl-positive cells, would significantly decrease the dose of each compound necessary to achieve a therapeutic effect. Here we demonstrate that bortezomib, in combination with the mitotic inhibitor paclitaxel, efficiently kill TKIs-resistant and sensitive Bcr-Abl-positive leukemic cells. In addition, bortezomib in combination with either paclitaxel or BI 2536, another mitotic inhibitor that inhibits PLK1, induces a marked downregulation of total and phosphorylated Bcr-Abl protein levels, thus downregulating the critical Bcr-Abl downstream signaling pathways and activating caspases. Similarly, bortezomib, in combination with other mitotic inhibitors, is able to decrease Bcr-Abl activity and increase caspase activation. Taken together, our findings unravel a novel promising treatment for TKIs-resistant and sensitive CML cases, as well as other Bcr-Abl positive leukemias. The effects of 9nM bortezomib, 6nM paclitaxel or both drugs in combination were evaluated in both parental K562 and TKIs-resistant K562-R cells after 48h of treatment. Similar to K562, K562-R cells are synergistically killed by bortezomib/paclitaxel regimen, as shown by a CI lower than 1. Notably, combined treatment with bortezomib and paclitaxel strongly decreases phosphorylation of Bcr-Abl, in both K562 and K562-R cell lines. The effects of 4nM bortezomib, 5nM paclitaxel or both drugs in combination for 48h were also evaluated in both parental LAMA84 and TKIs-resistant LAMA84-R cells. Interestingly, the LAMA84-R cells show a significant increase in total levels and phosphorylation of Bcr-Abl oncoprotein when compared with parental LAMA84 cells. This suggests that these cells adapted to resist imatinib, dasatinib and nilotinib treatments through the upregulation of Bcr-Abl levels and activity. Combined treatment with bortezomib and paclitaxel was able to downregulate total levels and phosphorylation of Bcr-Abl in LAMA84-R cell lines. The T315I point mutation in Bcr-Abl is known to confer resistance to imatinib, dasatinib, nilotinib, and bosutinib. It is
particularly sensitive when survival pathway inhibitors are combined with mitotic inhibitors. Moreover, combination of bortezomib with mitotic inhibitors are currently in clinical trials for the treatment of non-small-cell lung carcinoma and other solid tumors. Thus, we hypothesized that a strategy based on the combined treatment with bortezomib and mitotic inhibitors for the treatment of Bcr-Abl-positive leukemias may be promising. Especially important might be to determine the effectiveness of this strategy in TKIs-resistant Bcr-Abl-positive cases. Paclitaxel, a mitotic inhibitor drug acting by stabilization of microtubules, is FDA approved for the treatment of lung, ovarian, breast cancers and advanced forms of Kaposi’s sarcoma. Paclitaxel is now in clinical trials for the treatment of CML. However, to our knowledge, there are no clinical trials or published studies employing the combined bortezomib and paclitaxel regimen for the treatment of Bcr-Ablpositive CML. Such a combination, if synergistic in inducing apoptosis in Bcr-Abl-positive cells, would significantly decrease the dose of each compound necessary to achieve a therapeutic effect. Here we demonstrate that bortezomib, in combination with the mitotic inhibitor paclitaxel, efficiently kill TKIs-resistant and sensitive Bcr-Abl-positive leukemic cells. In addition, bortezomib in combination with either paclitaxel or BI 2536, another mitotic inhibitor that inhibits PLK1, induces a marked downregulation of total and phosphorylated Bcr-Abl protein levels, thus downregulating the critical Bcr-Abl downstream signaling pathways and activating caspases. Similarly, bortezomib, in combination with other mitotic inhibitors, is able to decrease Bcr-Abl activity and increase caspase activation. Taken together, our findings unravel a novel promising treatment for TKIs-resistant and sensitive CML cases, as well as other Bcr-Abl positive leukemias. The effects of 9nM bortezomib, 6nM paclitaxel or both drugs in combination were evaluated in both parental K562 and TKIs-resistant K562-R cells after 48h of treatment. Similar to K562, K562-R cells are synergistically killed by bortezomib/paclitaxel regimen, as shown by a CI lower than 1. Notably, combined treatment with bortezomib and paclitaxel strongly decreases phosphorylation of Bcr-Abl, in both K562 and K562-R cell lines. The effects of 4nM bortezomib, 5nM paclitaxel or both drugs in combination for 48h were also evaluated in both parental LAMA84 and TKIs-resistant LAMA84-R cells. Interestingly, the LAMA84-R cells show a significant increase in total levels and phosphorylation of Bcr-Abl oncoprotein when compared with parental LAMA84 cells. This suggests that these cells adapted to resist imatinib, dasatinib and nilotinib treatments through the upregulation of Bcr-Abl levels and activity. Combined treatment with bortezomib and paclitaxel was able to downregulate total levels and phosphorylation of Bcr-Abl in LAMA84-R cell lines. The T315I point mutation in Bcr-Abl is known to confer resistance to imatinib, dasatinib, nilotinib, and bosutinib. It is